Calcium Metal to Synthesize Amorphous or
Cryptocrystalline Calcium Phosphates
A. Cuneyt TAS, Ph.D.
Piscataway, New Jersey 08854, USA
E_mail: c_tas@hotmail.com www.cuneyttas.com
---------------------------------------------------------------------------------------------------------------------
Article: Mater. Sci. Eng. C, 32(5),
1097-1106 (2012)
US Patent: 9,108,860
B2 Patent issued on: August 18, 2015
---------------------------------------------------------------------------------------------------------------------
Metallic calcium was never used
before as the only calcium source in synthesizing bioceramics
in solutions mimicking the human blood plasma (in terms of their ion
concentrations). Amorphous calcium phosphate (ACP) powders were synthesized at
room temperature, in synthetic mineralization solutions which contained Na+, K+,
Mg2+, Cl-, HCO3- and HPO42-
ions at concentrations similar to those found in the human blood plasma, by using
calcium (Ca) metal as the only calcium source. The experimental conditions
leading to the formation of PCA (cryptocrystalline or poorly-crystallized
apatite) or CaCO3 powders were also determined when using metallic
Ca in aqueous synthesis in the mineralization solutions. The formation of
calcium phosphate (CaP) in synthesis solutions was
immediately initiated by the addition of calcium metal granules (or shots), at
appropriate amounts, into the solutions while the solutions were being
continuously stirred at room temperature (22±1°C). The
synthesis reactions were reaching completion in less than 30 minutes with the
final solution pH values ranging from 9 to 12, without a necessity for any
external pH adjustment in the form of any strong base (such as NH4OH,
LiOH, NaOH or KOH)
additions. ACP or PCA powders are useful for dentin and enamel
re-mineralization applications or orthopedic (bone) defect-filling
applications.
1.
Introduction
The systematic
synthesis and characterization of poorly-crystallized (i.e., cryptocrystalline)
apatite (PCA) powders in deionized (i.e., free of Na+, K+,
Mg2+, Cl- and HCO3- ions of blood
plasma) water solutions containing dissolved calcium nitrate tetrahdyrate (Ca(NO3)2×4H2O) and diammonium hydrogen phosphate ((NH4)2HPO4)
were initiated in the early 50’s by Hayek and co-workers [1-3]. The work of Hayek et al. [1-3] taught us to raise the pH values
of such cryptocrystalline apatite synthesis solutions to around 10.5-11 by the
addition of ammonium hydroxide (NH4OH). The method originally
developed by Hayek et al. [1-3] to produce cryptocrystalline nanoparticles of calcium
phosphate (CaP) was then adopted and popularized by Jarcho et al. [4]. Currently, the above-mentioned Hayek method of
synthesizing cryptocrystalline apatitic CaP powders is one of the most preferred.
Posner and
co-workers [5-7] were among the first, in the mid
50’s, to realize that the mineral of natural hard tissues consisted of
non-stoichiometric pseudoapatites. Posner et al. [5] envisaged in
as early as 1954 that the carbonate (CO3)- and foreign cation-free pseudoapatites can be represented by the formula of Ca10-xH2x(PO4)6(OH)2,
where the value of x would range from zero for stoichiometric hydroxyapatite
(HA) to two for an apatite with a Ca/P molar ratio of 1.333. One shall here
note the similarity between that pseudoapatite of
Ca/P ratio of 1.333 mentioned by Posner [5] and the
compound known as octacalcium phosphate (OCP, Ca8H2(PO4)6×5H2O or more
appropriately denoted as Ca8(HPO4)2(PO4)4×5H2O). Posner et al. [8] later showed
experimentally that the mineral of the rat bones did exhibit a very strong
age-dependent (from 1 to 40 days of age) variation in their Ca/P molar ratios,
i.e., the younger the rat the lower the Ca/P ratio of its bones, the very young
rats having a Ca/P ratio over the range of 1.15 to 1.34, in agreement with the
much earlier work of Burns and Henderson [9].
Posner and
co-workers [10] have been the first to describe
how to prepare synthetic amorphous calcium phosphate (ACP) powders by using
CaCl2- and (NH4)2HPO4-containing
distilled water solutions (i.e., not containing biologically essential ions
such as Na+, Mg2+, K+ or HCO3-)
whose pH values were raised to around 11 by NH4OH additions.
Betts and
Posner [11, 12] postulated that ACP actually
consisted of roughly spherical clusters (also called as Posner clusters) close to 1 nm in diameter, with a Ca/P molar ratio
of 1.5 and the formula of Ca9(PO4)6, which
were free of water. Synthetic ACP, according to Posner et al. [11, 12], consisted of roughly spherical Ca9(PO4)6 clusters, which
formed in water and were then aggregated randomly to produce the larger
spherical particles of ACP with the inter-cluster space being filled with
water. A similar aggregation process was however described previously, in 1957,
by Glimcher et
al. [13] in relation to mineralization in
the collagen-hydroxyapatite system.
ACP, when in
contact with an aqueous solution, is known to exhibit the unique ability to
first nucleate OCP-like nanosize crystallites on the
surfaces of its particles, which would then rapidly mature into apatitic calcium phosphate [10,
12]. This property of ACP powders
was successfully exploited to prepare injectable orthopedic cements [14, 15]. Posner and
his co-workers were also the first to study the interaction of casein micelles
of bovine milk with ACP powders [16],
and
this apparently led to the development of ACP-casein phosphopeptide
(CPP) [17]
complexes
for dental remineralization applications.
Since the
early studies of Hayek [1-3] and Posner [5, 10-13], to the best of our knowledge,
the ACP and PCA-related literature [18-35] did not contain any novel approaches to the synthesis
of ACP powders, i.e., meaning the calcium source employed in the synthesis
processes was always selected from the Ca-chloride, Ca-nitrate and Ca-acetate
salt group, and the pH values of the synthesis solutions were raised to the
basic range (pH ~11) by the
addition of strong bases such as NH4OH, NaOH
or KOH.
The current
study was originated by the following questions:
(i)
Could it ever be possible to synthesize CaP powders (either ACP or PCA) in aqueous solutions
totally free of nitrate (NO3), acetate (CH3COO) or
ammonium (NH4) ions which are not shown to be present in biological
bone or tooth formation processes?
(ii)
Could it be possible to synthesize ACP or PCA powders
by using aqueous solutions having the pH values from 9 to 12 (which was
underlined by the early works of Hayek and Posner as a necessity) without even
using the smallest aliquot of a strong base such as NH4OH, NaOH, KOH or LiOH?
(iii)
Could it be possible to simulate the concentrations of
inorganic ions present in human blood plasma in the synthesis solutions while
strictly maintaining the conditions set by the above two questions?
In CaP synthesis, nitrate or acetate ions would be introduced
into the synthesis solutions by the use of calcium nitrate tetrahydrate
or calcium acetate monohydrate as the calcium source. We considered that if we
were using Ca metal as the only calcium source, then that would totally
eliminate any nitrate or acetate ions. Layrolle et al. [27] used Ca metal
shots only to produce calcium diethoxide by reacting
them with ethanol, and they did not see the Ca metal shots as a possible
starting material to be quite useful in ACP or PCA synthesis in aqueous
solutions. It is common knowledge [36] that Ca metal
is produced by electrolysis of a molten bath of calcium chloride salt, and the produced
Ca metal granules react with distilled water to raise its pH under a slow
evolution of H2 gas (i.e., in
situ deprotonation). We considered that the use of Ca metal as the calcium
source in ACP or PCA synthesis might have eliminated the need for using of any
strong bases in raising the solution pH to the levels given in the works of
Hayek [1-3] and Posner [5, 10-13].
This study, to
the best of our knowledge, is the first one to use Ca metal (but not salts such
as Ca-chloride, Ca-nitrate or Ca-acetate) in synthesizing calcium phosphates.
This study also compared the use of Ca metal (as the Ca source) to using the
above-mentioned calcium salts. This study would also be the first one to
synthesize ACP (or PCA) powders in synthetic mineralization solutions developed
hereby to mimick the inorganic ion concentrations of
human blood plasma. Human body does not use deionized or distilled water in
synthesizing the mineralized portion of bone and teeth.
2.
Materials and Methods
2.1 Materials
Sodium
chloride (NaCl; Catalog No: 1.06404, Merck KGaA, Darmstadt, Germany), potassium chloride (KCl; Cat. No: 1.210517, Merck), magnesium
chloride hexahydrate (MgCl2∙6H2O; Cat. No: 459331,
Carlo Erba Reagenti, Milano, Italy), sodium
bicarbonate (NaHCO3; Cat. No: 1.06329, Merck), and disodium hydrogen phosphate
anhydrous (Na2HPO4; Cat. No: 1.06586, Merck) were used in
solution preparation.
Calcium metal
(Ca; spherical granular, 2-4 mm in diameter; Cat. No: 1.02053, Merck), calcium
chloride dihydrate (CaCl2∙2H2O; Cat. No: 1.02382,
Merck), calcium nitrate tetrahydrate (Ca(NO3)2×4H2O;
Cat. No: 1.02121, Merck), calcium acetate monohydrate (Ca(CH3CO2)2·H2O;
Cat. No: 402850, Sigma-Aldrich), and calcium hydroxide (Ca(OH)2;
Cat. No: 1.02110, Merck) were separately tested as the sources of calcium.
In some
experiments diammonium hydrogen phosphate ((NH4)2HPO4;
Cat. No: 1.01207, Merck) was tested as the source of phosphorus instead of Na2HPO4.
Finally, ammonium hydrogen carbonate (NH4HCO3, Cat. No:
1.01131, Merck) was also tested to replace NaHCO3 in some
experiments.
2.2 Solution preparation and synthesis
The solutions
were prepared in 500 mL-capacity PyrexÔ glass bottles (Fisher Scientific, Cat. No: FB800500).
The bottles were first cleaned by washing with 5 vol%
HCl, followed by rinsing with an ample amount of
doubly-distilled water, and overnight drying at 90°C. Five hundred mL of
doubly-distilled water was first placed into the bottles at room temperature
(RT, 22±1°C). A Teflon®-coated (25
mm long, 5 mm in diameter) rod-shaped magnetic stirrer was then placed into the
experiment bottle. All of the synthesis experiments were performed on a
magnetic stir-plate and the stirring rate for all experiments was kept constant
at 750 rpm. The carefully weighed chemicals were added, one by one, to the
bottle, under constant stirring of the solution inside. The next chemical was
not added prior to the complete dissolution of the previous one. Table 1 shows
the procedure of preparing the synthesis / mineralization solutions (MS) in 500
mL distilled water (not boiled prior to use to remove any possible HCO3-).
The chemicals were added to water in the order given. Table 1 offered three
choices of solution preparation to the reader; the first one would lead to
preparing a solution with 10 mM HPO42-,
whereas the third would result in a solution with 1 mM
HPO42-. All solutions shown in Table 1 were fully
transparent at the time of preparation, and thus they were ready for the
addition of the pre-weighed amount of Ca metal (or calcium chloride, calcium
acetate monohydrate or calcium nitrate tetrahydrate
in a limited number of experiments).
Table
1 Preparation of
mineralization solutions (MS) 500 mL H2O basis
Chemical g mM cation mM anion
KCl 0.1865 5 K+ 5 Cl-
MgCl2×6H2O 0.1525 1.5 Mg2+ 3 Cl-
NaCl 2.7760 95 Na+ 95 Cl-
NaHCO3 1.1341 27 Na+ 27 HCO3-
Choices:
(1)
Na2HPO4 0.7098 20 Na+ 10 HPO42-
(2)
Na2HPO4 0.3549 10 Na+ 5 HPO42-
(3) Na2HPO4 0.0710 2 Na+ 1 HPO42-
________________________________________________________________
To further
clarify the solution preparation technique described in Table 1; one first adds
KCl to 500 mL of water, dissolves it, then performs
the respective additions of MgCl2×6H2O, NaCl and
NaHCO3. At that moment, the solutions contain 5 mM
K+, 1.5 mM Mg2+, 103 mM Cl-, and 27 mM HCO3-.
These ion concentrations are identical with those of the blood plasma. If one
were then adding 0.7098 g of Na2HPO4, the solution would
have a total Na+ ion concentration equal to 142 mM.
This concentration of Na+ is exactly that of the blood plasma. The
solution thus obtained according to the choice-1 of Table 1 was able to match
the Na+, K+, Mg2+, HCO3-,
Cl- concentrations of the blood plasma, but will possess 10 times
the HPO42- concentration of plasma. However, the solution
of choice-3 (of Table 1) will have the identical HPO42-
concentration with that of blood plasma.
If one were
using CaCl2×2H2O
as the calcium source (instead of Ca metal), it would not be possible to
maintain the proper Cl- ion concentration in the solution, i.e., it
would have been in excess of 103 mM. Blood plasma
contains exactly 103 mM Cl-. If one were
using Ca(NO3)2×4H2O as the calcium
source, then the synthesis medium would have contained nitrate ions, which are
not present in the blood plasma. The same applies to the use of Ca-acetate, as
well.
Powder
synthesis began instantly by the addition of prescribed amount of calcium metal
granules into the mineralization solutions stirred at 750 rpm. Reactions were
continued for 25 minutes at RT (22±1°C). pH values were recorded (pH meter, Model: S40, Mettler-Toledo, w/combined pH-temperature electrode), at
every 30 seconds, starting from the moment of adding Ca metal into the
solutions. At the end of 25 minutes of stirring, the formed solids were
immediately and quickly filtered out of their mother liquors by using a Whatman No. 2 filter paper via a Buechner funnel apparatus, backed up
with a mechanical vacuum pump. The solid residues were washed with 750 mL of
distilled water and then dried on watch-glasses at RT for 48 hours in an
air-ventilated drying cabin. In the duplicate experiments, samples were
synthesized once more as described above, but then left in the solutions
overnight (i.e., at least 17 h), in the bottles, at RT. The pH values of the
solutions were measured once again after that long period of RT ageing and
exactly the same values were found with those measured after only 25 minutes of
reaction.
2.3 Sample characterization
Prior to
powder X-ray diffraction (XRD) and Fourier-transform infrared spectroscopy
(FTIR) analyses, the dried samples were ground, manually, in an agate mortar by
using an agate pestle. XRD runs were performed (Advance D8, Bruker, Karlsruhe,
Germany) in the step scan mode, with the step size of 0.02° and preset time of 5 seconds.
The powder diffractometer was equipped with a Cu tube and operated at 40 kV and
40 mA. XRD samples were prepared by gently packing the powders into the sample
holder cavity of around 1 mm-deep. FTIR samples were mixed with KBr powders at the ratio of 1 mg sample-to-250 mg KBr in an agate mortar. FTIR pellets of 13 mm diameter were
pressed at 10 tons. FTIR data were collected (Spectrum One, PerkinElmer,
Waltham, MA) by using 256 scans. Scanning electron microscopy (Vega-3, Tescan, A.S., Brno, Czech
Republic) samples were not ground and the small sample chunks were
sputter-coated with a thin gold layer before imaging.
3.
Results and Discussion
Until this
study, the researchers working in CaP-based
biomaterial synthesis have chosen either one of the following as their calcium
source; calcium chloride (anhydrous or dihydrate),
calcium nitrate (tetrahydrate), calcium acetate
(monohydrate), calcium carbonate or calcium hydroxide. The former three of
these have significant solubility even in cold water, but the latter two had
much lower solubilities in comparison to the former.
The major drawback associated with the use of calcium nitrate or calcium
acetate was that the synthesized phases would be poisoned by the residual
nitrate or acetate ions, as it was experimentally proven by Ivanova
et al. [37].
One of the
novelties of this study is that it used metallic calcium as the calcium source.
Metallic calcium presented clear advantages: (i) it did not bring into the
synthesis solutions any foreign or spectator anions, such as nitrates,
chlorides or acetates, and (ii) it
caused in situ deprotonation of the
aqueous synthesis media resulting in a smooth and rapid pH increase (as shown
below), totally eliminating the need for base (NaOH,
KOH, LiOH, NH4OH, etc.) additions to
maintain the synthesis pH above neutral. These two points further define the
novelty of this study. The necessity of maintaining the solution pH much above
the neutral during the synthesis of either ACP (amorphous calcium phosphate) or
PCA (poorly-crystallized, cryptocrystalline apatitic
calcium phosphate) was well-proven throughout the previous work of Hayek [1-3], Posner [5-7] and Rey [14, 15]. Rey et al. [15], for instance, prepared their
ACP powders by mixing an aqueous solution of Ca-nitrate with another solution
containing disodium hydrogen phosphate, 1 M NaOH and
0.3 M NaHCO3. The second solution [15] had a very
high Na+ concentration (i.e., 1.3 M). In stark contrast, the Na+
concentration of the solutions of the current study was kept constant at only
142 mM, which is the sodium concentration of human
blood plasma. Reaching pH values in the vicinity of 9 to 12 by using such low
concentrations of Na+ is another advantage of the use of Ca metal.
3.1 Synthesizing CaCO3 by using metallic Ca
The starting
point of this study was to find an answer to the following simple question:
what happens if one adds 25 mM (i.e., 10 times the
calcium concentration of blood plasma) of Ca metal granules (i) into water, (ii) into saline (NaCl-,
KCl- and/or MgCl2×6H2O-containing)
water, or (iii) into carbonated (HCO3—containing,
but no chlorides) water, and then stir the granules at RT in these solutions
for only a finite time, such as 25 minutes? The experiments detailed in Table 2
summarized the design of this study.
Calcium
granules stirred in doubly-distilled water for 25 minutes (with a rise in
solution pH to around 12) were not dissolved (Experiment-1 of Table 2), they
rather seemed to be rapidly covered with a white layer consisting of a biphasic
mixture of Ca(OH)2 and CaCO3, as determined by their XRD
data given in Figure 1a. XRD data, only in this case, were collected from the as-recovered granules, without
attempting to crush them. One can further speculate here that the incident
x-rays would not be able to pass through the hydroxide-carbonate layer formed
on the granules to reach their still metallic cores.
25 mM of calcium granules stirred for 25 minutes in an aqueous
solution containing only 5 mM K+, 1.5 mM Mg2+ and 27 mM HCO3-
did not totally dissolve. The Cl- ion concentration of this solution
was equal to 8 mM (Experiment-2 of Table 2), but the
K+ and Mg2+ concentrations were equal to that of blood
plasma. Ca granules did not dissolve in distilled water (Exp-1), and they also
did not totally dissolve in a solution containing 8 mM
Cl- and 27 mM HCO3-
(Exp-2). In these two experiments, the rapid formation of a biphasic layer of Ca(OH)2 (major phase) and CaCO3 (minor
phase) on the surfaces of the Ca metal granule was observed.
In Exp-3
(Table 2), 25 mM calcium granules were stirred in
distilled water containing only 27 mM Na+
and 27 mM HCO3- (no Cl-).
Granules did not dissolve. Very small amounts of solution precipitates formed
in experiments 2 and 3 proved, by XRD and FTIR, to be single-phase CaCO3.
Cl-
concentration was increased to 95 mM in Exp-4. 25 mM of calcium granules stirred in an aqueous solution
(Exp-4) containing 122 (=95+27) mM Na+, 95
mM Cl-, and 27 mM
HCO3- were dissolved completely and produced quite a
significant amount of CaCO3 precipitate in the solution in 25
minutes. We have thus experimentally determined that there seemed to be a close
relationship between the complete dissolution of the Ca metal granules and Cl-
concentration of the solution into which they were placed. Ca metal granules
added into aqueous solutions caused the evolution of H2 gas (i.e., in situ deprotonation [36]), but that
gas evolution slowed down by the formation of a hydroxide layer on the granule
surfaces at low Cl- concentrations. Moreover, since the granule size
used in this study was 2 to 4 mm, that gas evolution was not so fierce.
We speculate
that in solutions containing increased amounts of Cl-, H2
gas evolving at the granule surfaces was creating a microenvironment rich in HCl which could help to prevent the formation of the Ca(OH)2 layer, and with an increase in Cl-
concentration from 0 (Exp-1) to 8 (Exp-2), then to 95 mM
(Exp-4), the granules were dissolving in increasing amounts.
Experiment-5
was similar to experiment-4 but the MS solution (see Table 1) of Exp-5 also
contained K+ (5 mM), Mg2+ (1.5 mM), HCO3- (27 mM)
and Cl- (103 mM) ions at exactly the human
blood plasma levels. Half a gram of starting Ca granules was completely
dissolved and produced CaCO3 precipitates (1.228 g) at a high
process yield (98.15% of theoretical). The XRD data of the samples of experiments
4 and 5 (not shown) indicated CaCO3 of relatively high
crystallinity, individual XRD datum being indistinguishable from one another.
However, the FTIR data of CaCO3 produced in MS solution (Exp. 5) was
showing the O-H stretching vibration at around 3700 cm-1, as
indicated in Figure 1b. Based on observing the IR band at 1083 cm-1,
presence of very small amounts of vaterite may be
suspected, although XRD data did not show this phase. The photomicrographs of
the starting Ca granules and the calcite precipitated in Exp. 5 were given in
Figures 1c and 1d, respectively. The calcite crystals formed by adding 25 mM calcium granules into the MS solution (Exp. 5) had a
mean particle size of around 5 mm, exhibiting a high degree of agglomeration,
displayed nanosize steps and kinks reminding a
diffusion-controlled crystal growth kinetics on their surfaces, and by this
way, they differed from the clean and smooth-surfaced rhombic morphology of
calcite synthesized in distilled water as reported by Matijevic
et al. [38]. The first
five experiments (of Table-2) also helped to explain why an aqueous solution
with a Cl- concentration close to 100 mM
was needed for use with the Ca metal granules/shots. Human blood plasma
contains 103 mM Cl-, therefore, the
findings of the first five experiments were also indicating us the way to
develop a solution mimicking the ion concentrations of human blood plasma. The
MS solutions of this study are not SBF (synthetic body fluid) solutions since
they did not contain any Tris or Hepes,
which are not present at all in the human metabolism.
Figure 2
depicted the pH-time curves of the CaCO3 synthesis experiments by
using Ca granules. Ca metal granules were completely dissolved in experiments 4
and 5 at exactly the 11th minute. However, this specific time of
dissolution would surely depend on the stirring speed (750 rpm) employed, as
well as the volume and geometrical shape of glass bottles in which the
reactions were performed throughout this study.
All of the
above solutions and numbers may seem somewhat complicated at the first sight
but they actually point to a very simple fact, which could most probably be
explained by the below equations.
Ca(s) + H2O(l) ® Ca2+(aq) + 2OH- (aq) + H2 (g) (1)
Ca(s) + 2H2O(l) ® Ca(OH)2(s)
+ H2 (g) (2a)
Ca2+(aq) + HCO3- (aq) ® CaCO3(s)
+ H+ (aq) (2b)
Ca(OH)2(s) + H+ (aq) + Cl- (aq)
® Ca2+
(aq) + HCl + 2OH- (aq) (3).
Equation-1
explains the evolution of H2 gas and the observed rise in pH upon
adding the calcium granules into the solutions. Equations (2a) and (2b) explain
why the Ca granules did not dissolve in doubly-distilled water, and why the XRD
data of Figure 1 showed Ca(OH)2. Calcium
hydroxide, Ca(OH)2, is extremely prone to conversion at its surface
to calcite (CaCO3), and even many “pure,” commercial Ca(OH)2
powders have measurable amounts of CaCO3 in them, which can be
readily confirmed by a simple FTIR run to be performed on those so-called pure
and brand-new Ca(OH)2 samples. Equation 3 explains why the Ca-metal
granules readily dissolved in blood plasma-like, mineralization solutions (MS),
containing significant amounts (103 mM) of Cl- ions, in such a short time by causing
such a rapid rise in pH. There shall be a strong similarity between the
behavior of magnesium metal [39-43] and calcium metal in this
respect.
3.2 Synthesis of CaP in HCO3--free
solutions by using Ca metal
Ca metal
shots/granules were not expected to fully react in water only containing HPO42-
ions. In other words, in the absence of Cl- ions, the granules would
be easily covered with Ca-hydroxide and/or Ca-carbonate and would stop
reacting. This expectation was tested in experiment 6 (Table 2). 25 mM of calcium granules stirred in water only having 10 mM Na2HPO4 did not dissolve
completely, but the pH of the solution was able to rise above 12 and the small
amount of precipitates formed were found, by XRD (Figure 3a), to be comprised
of biphasic mixtures of cryptocrystalline apatitic CaP (PCA) and calcite.
Experiments 7
and 8 were performed to study the effect of Ca/P molar ratio, i.e., 1.667 and
2.50, in reacting Ca granules with the MS solutions free of HCO3-
ions. Both of these experiments produced cryptocrystalline apatitic
calcium phosphate (PCA) samples in solutions with final pH values greater than
12 (Figures 3a and 3b), without any calcite. It was important to notice the
characteristic stretching vibration of the O-H group at 3571 cm-1 in
the IR data (Fig. 3b) of the sample of Exp-8. Carbonates detected in the
samples of Figures 3a and 3b were due to the small amounts of dissolved HCO3-
present in the distilled water (not previously boiled) used. Calcium granules
reacted completely by the end of the 11th minute as shown in Figure
3c.
The MS
solutions of experiments 7 and 8 had 115 mM Na+,
103 mM Cl-, 5 mM
K+, 1.5 mM Mg2+ and 10 mM HPO42-, and in both experiments
one would be able to freely change the Ca content without disturbing the
concentration of any other ion in the solution; i.e., another advantage of
using Ca metal in CaP synthesis. This would not be
possible if one were using, for instance, CaCl2×2H2O as the calcium
source.
Experiments 7
and 8, therefore, showed a simple way of producing cryptocrystalline (some call
it poorly-crystalline or poorly-crystallized or nanocrystalline)
apatitic CaP powders at RT,
in a very short 25 minutes, without employing any external pH control technique
(such as drop-wise addition of a strong base such as NH4OH, NaOH, KOH,or
LiOH) at an in
situ solution pH of 12. Exp-8 had the nominal, solution Ca/P molar ratio of
2.5, which was equal to that of blood plasma. Bacteria cannot grow at a
solution pH of 12, but they definitely can if the synthesis solutions were at
neutral pH (6.8 to 7.6). This is another advantage of using Ca metal in PCA
synthesis.
Experiments 9
and 10 (Table 2) were replacing the Na2HPO4 used in
experiments 7 and 8 with (NH4)2HPO4, while
keeping all the other synthesis parameters unchanged. Although the presence of
NH4 ions in a synthesis system claiming to mimic the ions and ion
concentrations in blood plasma would not be acceptable, experiment 9 produced
amorphous calcium phosphate (ACP) at the Ca/P molar ratio of 1.667 and the
final pH value of 11.3.
It was quite
easy to distinguish between the ACP and PCA phases by using their FTIR data, as
exemplified by the IR traces of experiments 9 and 7 in Figure 3b, respectively.
In the IR data of ACP samples the phosphate bands over the range of 660 to 490
cm-1 do not show that splitting, which was otherwise observed in PCA
samples. When the Ca/P molar ratio was increased to 2.5 in experiment 10, the
produced powders were not ACP but PCA. The solution pH in this experiment was
12. Upon repeating the experiment 9, but ageing the formed precipitates in the
mother solution for 5 days at RT (solution pH dropping to 10.7 from 11.3, in 5
days), followed by filtering and drying, the obtained powders were consisted of
PCA, not ACP, as shown in Figure 3a. This was quite an expected result since
ACP was not a stable phase (even in its mother liquor over a period of 5 days)
and it acted as a precursor to PCA, as previously shown by Cazalbou
et al. [44].
3.3 Synthesis of CaP in HCO3--free
solutions by using CaCl2×2H2O instead of Ca metal
Upon replacing
the Ca metal with CaCl2×2H2O, the pH values of synthesis solutions
drastically suffered from this change. Experiment 11 in comparison to
experiment 6 showed that drastic drop in solution pH from 12.3 to 5.9. At such
a low pH (5.9), it was inevitable to form DCPD (dicalcium
phosphate dihydrate; brushite;
CaHPO4×2H2O).
The comparison of the XRD and FTIR data of experiments 6 (with Ca metal in
water) and 11 (with Ca-chloride in water) was given in Figure 4.
Experiments 12
through 15 tested the formation of calcium phosphates in water and HCO3--free
MS solutions by using Ca-chloride and diammonium
hydrogen phosphate as the starting chemicals. In these experiments solution pH
values remained between 5.7 and 6.5, and the obtained precipitates contained
DCPD as the major phase.
If one used
Ca-chloride dihydrate instead of Ca metal, as the
calcium source, to synthesize CaP in HCO3-
-free plasma-like solutions, mildly acidic DCPD would be the major phase
obtained. The
readers shall compare the synthesis conditions and the results of experiments
8, 10 and 13 with one another.
3.4 Synthesis of ACP in MS solutions by using Ca metal
Experiments 16
through 18 tested the synthesis conditions closest to the ionic concentrations
of the human blood plasma, by using the metallic Ca granules. In experiment 16;
calcium, phosphate (HPO42-), bicarbonate (HCO3-),
potassium, chloride, and magnesium ion concentrations were made identical with
that of blood plasma, but in that experiment the sodium concentration was equal
to 124 mM. In experiment 18, on the other hand;
bicarbonate (27 mM), sodium (142 mM),
magnesium (1.5 mM), potassium (5 mM)
and chloride (103 mM) ion concentrations were
identical with that of blood plasma. In other words, experiments 16 through 18
tested the MS solutions given in Table 1 under three different choices. The
combined XRD and FTIR data of the resultant ACP samples were given in Figure
5a. The second
inset of Figure 5a confirmed the absence of the octacalcium
phosphate (OCP, Ca8(HPO4)2(PO4)4×5H2O) phase in the samples
of experiments 16 to 18. At such high solution pH values it would be very
difficult, if not impossible at all, to observe acidic OCP. The sample of
experiment 18 showed the presence of a small amount of calcite (CaCO3)
phase in its XRD data. However, when we duplicated experiments 16 through 18,
and left the precipitate-containing solutions overnight without stirring,
followed by filtering and drying, the resultant XRD data of especially
experiment 18 did not show that second phase of calcite. All three samples (16
through 18) depicted the characteristic XRD pattern of ACP. The Ca metal
granules in experiments 16 through 18 all dissolved/disappeared at around the 11th
minute. When experiment 18 is performed (i.e., experiment 19) in
doubly-distilled water (containing 10 mM HPO42-,
27 mM HCO3-, and 47 mM Na+), instead of the MS solution, Ca metal
granules did not dissolve and no precipitates were obtained. This again proved
the role of Cl- ions, as explained by equations (1) through (3)
above.
Figure 5b
showed the pH-time curves for experiments 16 through 19. The curves for
experiments 16 through 18 in this figure, as well as the previous pH-time
curves (Fig. 3c), exhibited a nonlinear increase of pH in a time dependent
manner and they were approximated (TableCurve, v1.10,
Jandel Scientific, 1993) by the logistic
dose-response function (y = a + [b / (1 + (x/c)d)]), for which the experimental
parameters were given below in Table 3.
The SEM
photomicrographs of samples obtained from experiments 16 and 18, were given in
Figures 5c and 5d, respectively. It should be noted that these are filtered and
dried samples, they were not even lyophilized upon separation from their mother
liquors. Regular drying causes agglomeration of individual particles or
moieties.
Table 3 Results of logistic dose-response
curve fitting on the pH-time curves
__________________________________________________________________________
Parameters Exp 16 Exp 17 Exp
18 Exp 8 Exp 5
a 8.2495 8.3653 8.4338 12.6960 8.4163
b 0.9421 2.0039 3.5657 -3.7066 3.9064
c 0.7841 0.7827 1.3204 0.0222 1.4376
d -1.9020 -1.3032 -1.7912 0.4083 -1.8735
r2 0.9994 0.9985 0.9936 0.9794 0.9922
Fit
Std Error 0.0043 0.0139 0.0608 0.0739 0.0760
_________________________________________________________________________
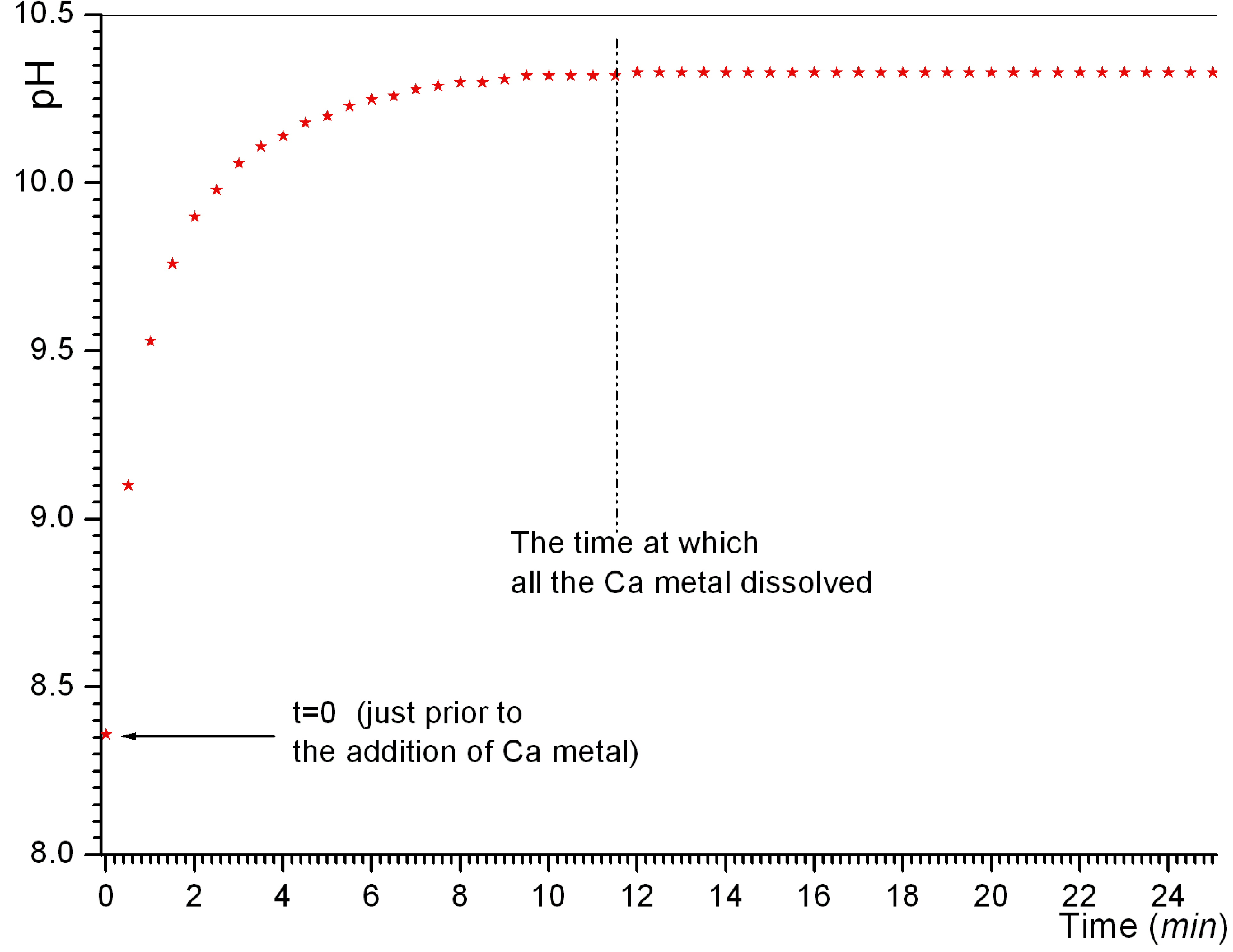
Link to a characteristic plot of solution pH versus time → http://www.cuneyttas.com/image001.jpg
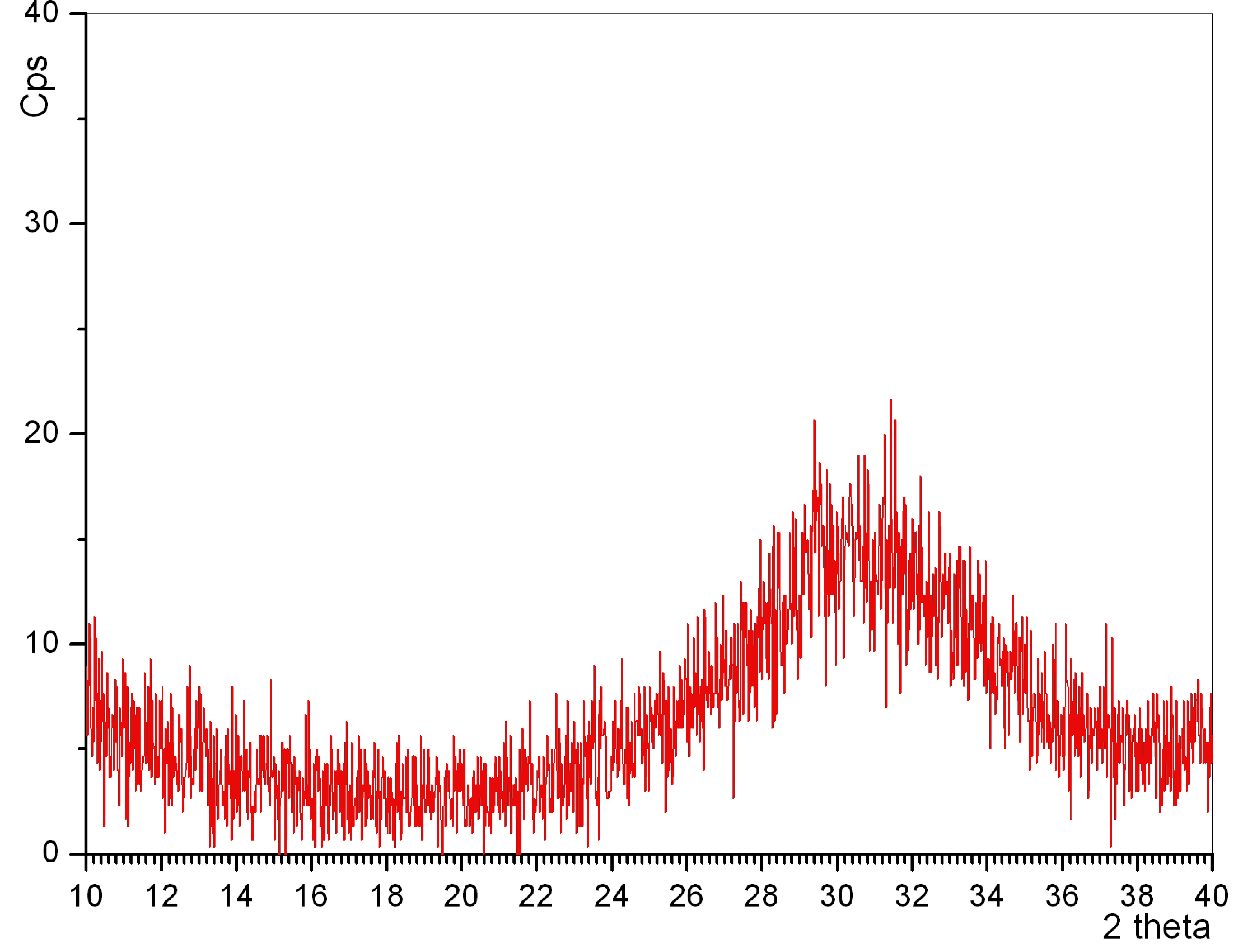
Nevertheless, it was apparent
from Figures 5c and 5d that the average particle diameter in these x-ray
amorphous, carbonated and mesoporous CaP powders was
pretty much less than 70 nm. This is the particle size directly observed by the SEM,
not the crystallite size. Crystallite sizes cannot be determined by using the Scherrer equation while using the XRD data of x-ray
amorphous samples (Fig. 5a).
Link to a typical XRD plot of ACP
powders → http://www.cuneyttas.com/image002.jpg
The
concentration of Ca metal added into the MS solutions (starting from 2.5 mM in experiment 16 and going up to 25 mM
in experiment 18) was found to be quite influential on the final pH values
attained in syntheses. When the Ca concentration was kept equal to that of the
blood plasma (i.e., 2.5 mM in exp. 16), the pH of the
solution has risen only to 9.2 and stabilized at that value. By increasing it
to 12.5 mM (i.e., 5 times that of plasma in exp. 17),
the pH rose to 10.3, and the pH increased to 12 when the Ca concentration in
the MS solution was increased to ten times that of the blood plasma (exp. 18).
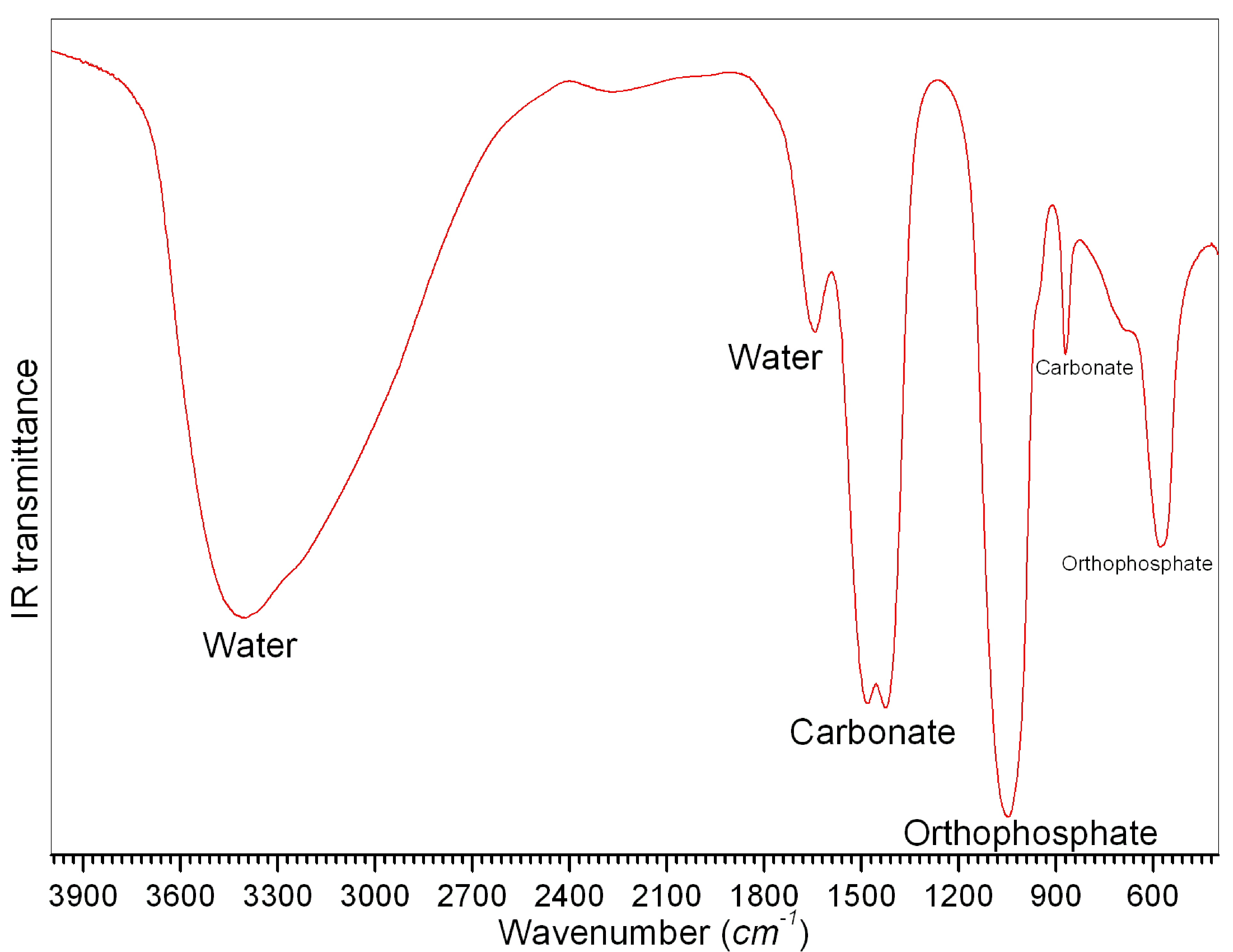
Link to a characteristic FTIR
plot of the ACP powders obtained by using Ca metal → http://www.cuneyttas.com/image003.jpg
The conditions
of Exp-16 was of pivotal significance for this study, since the Ca2+,
HPO42-, HCO3-, Mg2+, K+,
Cl- concentrations of this experiment were identical with those of
human blood, and moreover, no foreign ions such as nitrate, ammonium and
acetate were introduced to the synthesis process. As shown by the data of Fig.
5b, maintaining a literally constant pH in CaP
synthesis, without employing any pH control (such as adding bases or acids to
keep the pH constant), was never shown before to be possible. These define the
novelty and practicality of the approach of using Ca metal as the sole calcium
source in CaP synthesis.
Link to the SEM photomicrograph
of nanosize ACP spheres obtained by using Ca metal → http://www.cuneyttas.com/image004.jpg
3.5 Synthesis of ACP in MS solutions by using Ca metal, ammonium phosphates
and ammonium carbonate
The
influence of the use of (NH4)2HPO4 and NH4HCO3
salts, instead of Na2HPO4 and NaHCO3 was also
tested in synthesizing CaP powders by using Ca metal
granules. Such a direct comparison was necessary. Experiments 20 through 24 (of
Table 2) all produced ACP powders in MS solutions. The use of Na-phosphate or
Na-bicarbonate (as shown in experiments 20 and 21) kept the solution pH at
above 10, but when both of Na2HPO4 and NaHCO3
were replaced by (NH4)2HPO4 and NH4HCO3
the solution pH values dropped to about 9.3 to 9.5 (experiments 22 through 24).
Of course, the solutions used in these experiments could not mimic the
physiological solutions, since they contained significant amounts of ammonium
ions which are not found in blood plasma. The XRD and FTIR data of experiments
22 through 24 were shown in Figure 6. However, the direct comparison of Exp-18
and Exp-24 would yield that it would be possible to produce carbonated ACP
powders, by using Ca metal, at pH values of 12 and 9.5, respectively, without
using any external pH adjustment controls.
3.6 Synthesis of PCA in MS solutions at pH 7 without using Ca metal
Experiments
25 through 30 of Table 2 studied the synthesis of CaP
in MS solutions, without using Ca metal. These experiments were planned to show
what difference the use of Ca metal would really cause in comparison to the
more commonly preferred calcium ion sources, such as CaCl2×2H2O, calcium acetate
monohydrate (Ca(CH3CO2)2·H2O),
Ca(NO3)2×4H2O, and Ca(OH)2. Figure 7 showed the XRD traces
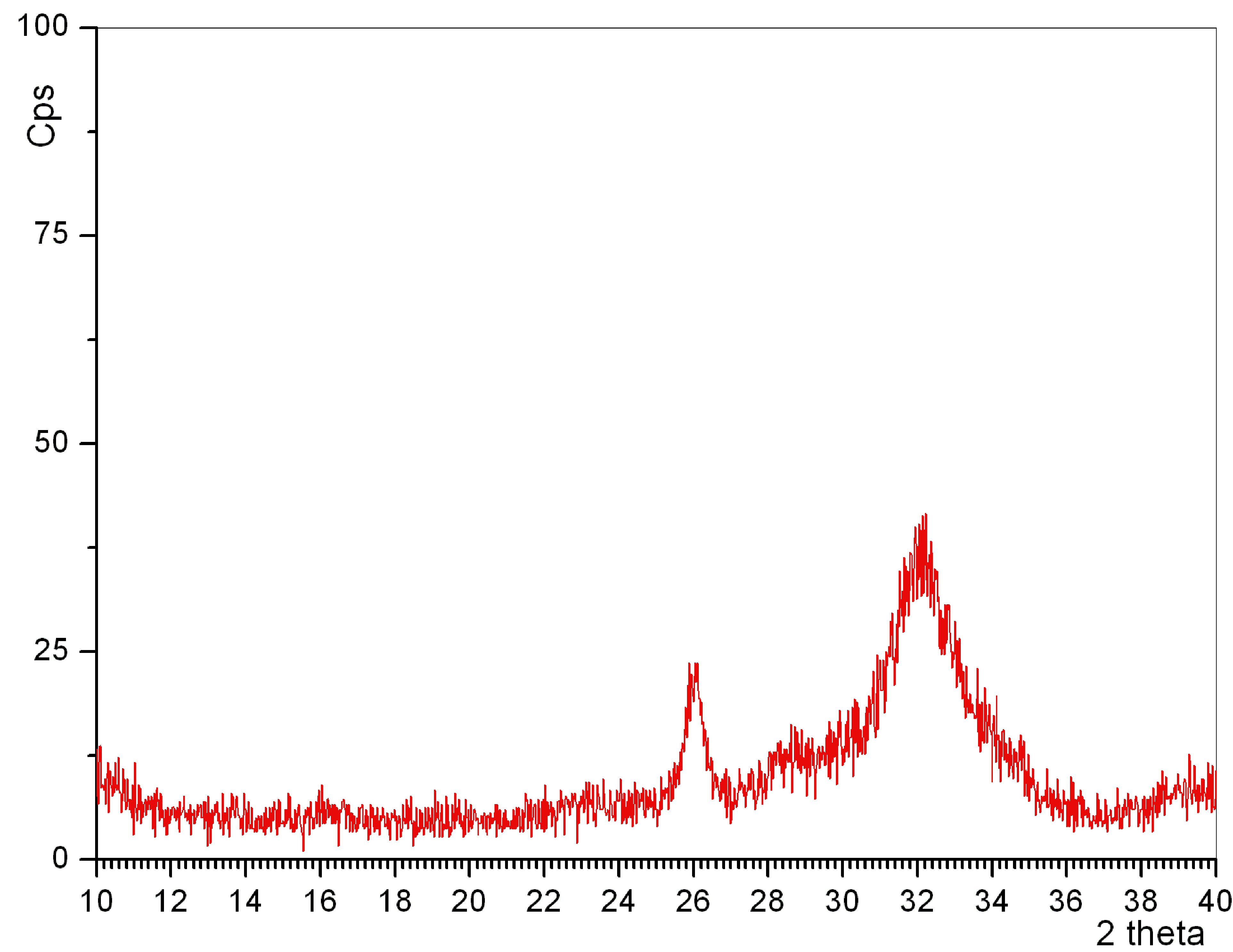
of samples obtained in experiments 25 through 29, all indicating PCA. The inset
in Figure 7, on the other hand, exhibited the IR traces of the samples of
experiments 25 through 27. The IR traces of experiments 26, 28 and 29 were very
similar to one another, and they all exhibited much less carbonate ion presence
(according to the qualitative IR data) in comparison to, for instance, the
sample of experiment 27.
Link to the characteristic XRD plot of
PCA powders synthesized by using Ca metal → http://www.cuneyttas.com/image005.jpg
MS solutions
were working perfectly well, at the stated ion concentrations, in providing a
reaction pH of exactly 7.0 for Ca-chloride, Ca-acetate, or Ca-nitrate; without
a need for any external pH adjustments by acids or bases of any kind. Ca metal
granules made it possible to synthesize ACP or PCA powders at pH values higher
than 7.0, without needing any base additions for pH control, in MS solutions.
Link to the typical FTIR plot of
PCA powders → http://www.cuneyttas.com/image006.jpg
To synthesize
PCA by using Ca metal granules, we found that one needed to eliminate HCO3-
from the MS solutions. Using CaCl2×2H2O in doubly-distilled water or HCO3--free
MS solutions containing phosphate ions, without any pH adjustments, would never
allow the synthesis of PCA, since the pH of the solutions was lower than
neutral (i.e., 7) and would thus only be suitable for the crystallization of brushite (CaHPO4×2H2O) phase, as also shown in this study.
Links to characteristic SEM
photomicrographs of PCA powders synthesized by using Ca metal → http://www.cuneyttas.com/image007.jpg
http://www.cuneyttas.com/image008.jpg
http://www.cuneyttas.com/image009.jpg
3.7 Ca metal granules or Ca(OH)2 in MS
solutions?
XRD and FTIR analysis
of the sample obtained in experiment 30 (Table 2), which opted for 25 mM Ca(OH)2 to be added into the typical MS
solution of this study, tried to provide an answer to the question of this
section. Figure 8a compared the XRD traces of all the samples of this study
which comprised of a biphasic mixture of ACP and CaCO3 after 25
minutes of stirring at RT in the MS solutions. The main comparison should
actually be made between the sample 18 (25 mM Ca) and
sample 30 (25 mM Ca(OH)2)
in the chart of Figure 8a, since ammonium ions were present in the solutions of
sample 20 and 21. Solution-wise, samples 20 and 21 do not compare well with
those of samples 18 and 30. When Ca metal in experiment 18 was replaced by Ca(OH)2 in experiment 30, while keeping all the
other synthesis parameters constant, the amount of the secondary phase of CaCO3
significantly increased (Fig. 8a). The FTIR data of the same experiments were
given in Figure 8b. Figure 8b provided the evidence that the sample of
experiment 30 was also poisoned with unreacted Ca(OH)2,
i.e., presence of the Ca(OH)2-specific IR band recorded at around
3650 cm-1. Moreover, the sample of experiment 30 showed the
characteristic IR bands of the calcite phase at 2513, 1798, 875 and 712 cm-1.
In the duplicate experiments same results were obtained meaning that
Ca-hydroxide was not able to completely react to form ACP in the MS solutions
by fully consuming itself.
3.8 Significance of synthesizing CaP in
mineralization solutions free of Tris or Hepes
Human blood, which
provides the necessary nutrients to the trabecular/cancellous bones and the
dentine of teeth, does not contain Tris (or Hepes), nitrate, acetate and/or ammonium ions. Therefore,
it would be difficult to classify the synthesis (or coating) processes using Tris-HCl (or Hepes-NaOH) buffered
solutions and especially the synthesis methods using one or more of the
starting chemicals of Ca-nitrate tetrahydrate,
Ca-acetate monohydrate, ammonium hydroxide, diammonium
hydrogen phosphate or ammonium dihydrogen phosphate as properly mimicking the physiological
processes [45-49].
Ammonium-,
nitrate- and acetate-free synthesis recipes (especially those of experiments 7,
8, 16, 17 and 18) given in Table 2 of this study provided easy-to-reproduce and
quite simple procedures to synthesize PCA (cryptocrystalline apatitic CaP) and ACP (x-ray
amorphous CaP) powders at RT in glass media bottles,
without requiring special reactor designs and pH adjustment/control measures.
It would be naïve to assume that the PCA or ACP synthesized in such blood
plasma-like solutions would be free of ionic substitutions of Na+, K+,
Mg2+, CO32- and Cl- ions at the
crystallographic Ca, PO4 and OH sites of hydroxyapatite structure.
In a follow up study, we will publish the results of ICP-AES (inductively-coupled
plasma atomic emission spectroscopy) analyses on such samples in comparison to
PCA or ACP synthesized in synthesis media free of K+, Mg2+
and Cl- ions.
The ionic
strength of the synthesis solutions (after the addition of Ca metal granules)
of experiments 7, 8, 16, 17 and 18 of this study was adjusted to be 167.83,
184.5, 139.5, 171.5 and 211.5 mM, respectively. If
one were to prepare an aqueous solution comprising 2.5 mM
Ca2+, 1 mM HPO42-,
142 mM Na+, 5 mM
K+, 1.5 mM Mg2+, 27 mM HCO3- and 103 mM
Cl- (i.e., the exact ion concentrations of human blood plasma) then
the ionic strength of that solution would have been 148.5 mM.
The ionic strengths higher than 148.5 mM were
intentionally chosen in this study to facilitate the synthesis of larger
amounts of PCA or ACP powders.
The influence
of synthesis pH on the CaP formation seemed to be not
receiving the required attention in the previous literature. To the best of our
knowledge, there are very few studies to mention the basicity of apatitic CaP forming in solutions
with pH values around 11. The current study obtained pH values from 9 to 12
without adding any base. Liu et al. [50] used the Ca-nitrate/(NH4)2HPO4
route and studied the ACP and apatitic CaP precipitation at pH 10 to 11, whereas the high pH
values in that study were apparently obtained by NH4OH additions.
Liu et al. [50] study was not designed to measure the basicity of the
CaP formed. The lack of previous studies on the
basicity of apatitic CaP
may even force the field researchers to think that apatite (which is basically
a hydroxyl-containing phosphate in its formula and structure) is not a compound
with a significantly basic surface, which is not true. However, the work of Tsuchida et al. [51] deliberately and quantitatively studied the surface
basicity of apatitic Ca/P, by again using the
Ca-nitrate//(NH4)2HPO4
route of synthesis (with ammonia additions during synthesis) and found that (i) the solution
pH had the greatest influence on the Ca/P ratio of apatitic
CaP produced and (ii)
the basic site density in apatite depended only on the Ca/P ratio of the
sample. Therefore, the current study using Ca metal provided a very simple
method of synthesizing CaP at the high pH values
(from 10 to 12) studied separately by Liu et
al. [50] and Tsuchida
et al. [51].
Most of our
samples produced at pH values 9 to 12 were of poor crystallinity. Nelson [52] investigated the reason for the poor crystallinity of
Na+- and CO32--containing apatitic
CaP samples (usually prepared by high temperature (90°<T<250°C) processes) by using TEM and
found out that the reason was not a decrease in overall particle size but the
fact that each particle consisted of agglomerates of small crystalline domains
each having a different orientation. The domain sizes appeared to decrease with
an increase in the carbonate content and could become as small as 8 nm. Nelson [52] also found out that the simultaneous incorporation of
Na+ and CO32- ions into the apatitic CaPs resulted in an
increased rate of dissolution for the solid. The formula for the Na- and CO3-doped
CaP could be as complex as Ca10-xNax[(PO4)6-x(CO3)4x/3][(OH)2-2x/3], where 0 £ x £ 3 [53]. A similar formula for K- and CO3-doped CaP shall be expected. Nonstoichiometric CaP phases would usually have slightly higher aqueous
solubility with respect to their perfectly stoichiometric counterparts [54].
Although the
size of the Mg2+ ion (0.066 nm) is quite smaller than that of Ca2+
(0.101 nm), magnesium ions can substitute for Ca in a number of CaP phases, including whitlockite
(Ca3(PO4)2) [55]. The incorporation of Mg into amorphous CaP has been relatively well studied. Termine
et al. [56] found that
the elapsed time between the precipitation of ACP and its solution-mediated
transformation into cryptocrystalline apatitic CaP (PCA) may be increased considerably with the addition
of small amounts of Mg2+ ions. The current study was not focused on
the hydrothermal transformation of ACP into PCA or vice versa, however our
synthesis solutions (MS) contained Mg2+.
The author’s
lab has been the first to synthesize cryptocrystalline apatitic
CaP powders in Tris-buffered
SBF (synthetic body fluid) solutions (by using Ca-nitrate) at 37°C and to show (via ICP analyses)
that Mg and Na were indeed incorporated into the obtained powders [46]. Such biomimetic apatite powders were also shown to
possess unprecedented high stability against thermal decomposition [46].
For readers
who may ask the question of why one would need a solution pH as high as 9.2 (as
in Exp-16) to synthesize CaP mimicking the
physiological processes, it is a well-known fact that alkaline phosphatase
(ALP) enzyme is secreted in bones by the osteoblast cells while depositing nanosize apatitic CaP crystals, and the optimum pH of ALP secretion is
between 9.5 and 10.5 [57-59]. Synthesis procedures described
for Experiments 16 and 17 in Table 2 were both able to produce the ACP phase at
or around this biomimetic pH value of ALP secretion.
It was also
shown in this study, in contrast to some previous reports, that the use of
synthetic polymers, which do not have any place in the human metabolism, was
not necessary at all to synthesize ACP in aqueous media [60].
Magnesium
metal was already tested [39-43] as a starting material for biomedical
scaffolds, the current study may initiate the use of Ca metal for the same
purpose.
4.
Conclusions
Metallic
calcium was used for the first time in synthesizing CaCO3,
poorly-crystalline (cryptocrystalline) apatite (PCA) or x-ray amorphous calcium
phosphate (ACP) powders.
Calcium
phosphate synthesis with metallic Ca was tested both in doubly-distilled water
and in water containing ions found in human blood.
The use of
metallic Ca eliminated the need for external pH control in calcium phosphate
synthesis solutions in the form of adding strong bases such as NaOH, KOH, LiOH or NH4OH.
The use of
metallic Ca made it possible to synthesize PCA or ACP powders in solutions
completely free of foreign ions such as ammonium, nitrate or acetate, which are
not encountered in human blood.
Notes
Certain commercial equipments, instruments, or chemicals are only
identified in this paper to foster understanding. Such identification does not
imply recommendation or endorsement by the author, nor does it imply that the
equipment or materials identified are necessarily the best available for the
purpose.
References
[1] E. Hayek, F. Mullner, and K. Koller, “Zur Kenntnis des Hydroxylapatits,” Monatsh. Chem. 82 (1951) 958-969.
[2] E. Hayek,
J. Lechleitner, and W. Bohler,
“Hydrothermal Synthese von Hydroxylapatit,”
Angew. Chem. Int. Edit. 67 (1955) 326-326.
[3] E. Hayek and H. Newesely,
“Pentacalcium Hydroxyorthophosphate,”
Inorganic Syntheses, Volume VII, pp.
63-65. McGraw-Hill, Inc., 1963.
[4] M. Jarcho, C.H. Bolen, M.B. Thomas, J. Bobick,
J.F. Kay, and R.H. Doremus, “Hydroxylapatite
Synthesis and Characterization in Dense Polycrystalline Form,” J. Mater. Sci. 11 (1976) 2027-2035.
[5] A.S.
Posner, C. Fabry, and M.J. Dallemagne,
“Defect Apatite Series in Synthetic and Natural Calcium Phosphates: The Concept
of Pseudoapatites,” Biochim. Biophys. Acta
15 (1954) 304-305.
[6] A.S.
Posner, J.M. Stutman, E.R. Lippincott,
“Hydrogen-bonding in Calcium-deficient Hydroxyapatites,” Nature 188 (1960) 486-487.
[7] M.I. Kay,
R.A. Young, and A.S. Posner, “Crystal Structure of Hydroxyapatite,” Nature 204 (1964) 1050-1050.
[8] A.S.
Posner, R.A. Harper, S.A. Muller, and J. Menczel “Age
Changes in the Crystal Chemistry of Bone Apatite,” Ann. NY. Acad. Sci. 131 (1965) 737-742.
[9] C.M.
Burns and N. Henderson, “Influence of Age on the Mineral Constituents of Bones
from Pups and Kittens,” Biochem. J. 30 (1936) 1207-1213.
[10] N.C.
Blumenthal, J.M. Holmes, and A.S. Posner, “Effect of Preparation Conditions on
the Properties and Transformation of Amorphous Calcium Phosphate,” Mater. Res. Bull. 7 (1972) 1181-1190.
[11] F. Betts
and A.S. Posner, “An X-ray Radial Distribution Study of Amorphous Calcium
Phosphate,” Mater. Res. Bull. 9
(1974) 353-360.
[12] A.S.
Posner and F. Betts, “Synthetic Amorphous Calcium Phosphate and Its Relation to
Bone Mineral Structure,” Acc. Chem. Res.
8 (1975) 273-281.
[13] M.J. Glimcher, A.J. Hodge, and F.O. Schmitt, “Macromolecular
Aggregation States in Relation to Mineralization: The Collagen-Hydroxyapatite
System as Studied in vitro,” P. Natl. Acad. Sci. USA 43 (1957) 860-867.
[14] A. Tofighi, S. Mounic, P. Chakravarthy, C. Rey, and D. Lee, “Setting Reactions
Involved in Injectable Cements based on Amorphous Calcium Phosphate,” Key Eng. Mat. 192-1 (2000) 769.
[15] D.D. Lee,
C.Rey, M. Aiolova, and A. Tofighi, “Method of Preparing a Poorly Crystalline Calcium
Phosphate and Methods of Its Use,” U.S. Patent No: 7,517,539 April 14, 2009.
[16] T.C.A. McGann, R.D. Kearney, W. Buchheim,
A.S. Posner, F. Betts, and N.C. Blumenthal, “Amorphous Calcium Phosphate in
Casein Micelles of Bovine Milk,” Calcified
Tissue Int. 35 (1983) 821.
[17] M.
Bannon, R.H. Hammond, and E.C. Reynolds, “Amorphous Calcium Phosphate-Casein Phosphopeptide (ACP-CPP) as a Dentinal Hypersensitivity Treatment
Agent,” J. Dent. Res. 74 (1995) 754.
[18] H. Fleisch, R.G.G. Russell, S. Bisaz,
J.D. Termine, and A.S. Posner, “Influence of
Pyrophosphate on Transformation of Amorphous to Crystalline Calcium Phosphate,”
Calc. Tiss.
Res. 2 (1968) 49.
[19] E.D. Eanes, “Thermochemical Studies on Amorphous Calcium
Phosphate,” Calc. Tiss.
Res. 5 (1970) 133.
[20] A.L. Boskey and A.S. Posner, “Magnesium Stabilization of
Amorphous Calcium Phosphate -Kinetic Study,” J. Dent. Res. 52 (1973) 167.
[21] R.Z. LeGeros, W.P Shirra, M.A. Miravite, J.P. LeGeros,
“Biological and Synthetic Amorphous Calcium Phosphates,” J. Dent. Res. 53 (1974) 117.
[22] M.J. Glimcher, L.C. Bonar, M.D. Grynpas,
W.J. Landis, A.H. Roufosse, “Recent Studies of Bone
Mineral – Is the Amorphous Calcium Theory Valid,” J. Cryst. Growth 53 (1981) 100.
[23] M.S. Tung
and W.E. Brown, “An Intermediate State in Hydrolysis of Amorphous Calcium
Phosphate,” Calcified Tissue Int. 35
(1983) 783.
[24] H.A. Lowenstam and S. Weiner, “Transformation of Amorphous
Calcium Phosphate to Crystalline Dahllite in the Radular Teeth of Chitons,” Science 227 (1985) 51.
[25] L. Brecevic, V. Hlady, and H. Furedi-Milhofer, “Influence of Gelatin
on the Precipitation of Amorphous Calcium Phosphate,” Colloid. Surface. 28 (1987) 301.
[26] D. Skrtic, E.D. Eanes, and J.M. Antonucci, “Dissolution Behavior
of Amorphous Calcium Phosphate Methacrylate Composites,” J. Dent. Res. 73 (1994) 302.
[27] P. Layrolle, A. Ito, and T. Tateishi,
“Sol-gel Synthesis of Amorphous Calcium Phosphate and Sintering into
Microporous Hydroxyapatite Bioceramics,” J. Am. Ceram. Soc. 81 (1998) 1421.
[28] A.
Rodrigues and A. Lebugle, “Behavior
in Wet Atmosphere of an Amorphous Calcium Phosphate with an Atomic Ca/P Ratio
of 1.33,” J. Solid State Chem. 148
(1999) 308.
[29] M. Kazanci, P. Fratzl, K. Klaushofer, E.P. Paschalis, “Complementary Information on
In Vitro Conversion of Amorphous (precursor) Calcium Phosphate to
Hydroxyapatite from Raman Microspectroscopy and
Wide-angle X-ray Scattering,” Calcified
Tissue Int. 79 (2006) 354.
[30] T. Tsuji,
K. Onuma, A. Yamamoto, M. Iijima,
and K. Shiba, “Direct Transformation for Amorphous to
Crystalline Calcium Phosphate facilitated by Motif-programmed Artificial
Proteins,” P. Natl. Acad. Sci. USA
105 (2008) 16866.
[31] Z.Z. Zyman, D.V. Rokhmistrov, and V.I.
Glushko, “Structural and Compositional Features of
Amorphous Calcium Phosphate at the Early Stage of Precipitation,” J. Mater. Sci. Mater. M. 21 (2010) 123.
[32] H.H. Pan,
X.Y. Liu, R.K. Tang, and H.Y. Xu, “Mystery of the Transformation from Amorphous
Calcium Phosphate to Hydroxyapatite,” Chem.
Commun. 46 (2010) 7415.
[33] D. Rabadjieva, R. Gergulova, R. Titorenkova, S. Tepavitcharova,
E. Dyulgerova, C. Balarew,
and O. Petrov, “Biomimetic Transformations of
Amorphous Calcium Phosphate: Kinetic and Thermodynamic Studies,” J. Mater. Sci. Mater. M. 21 (2010) 2501.
[34] D. Lee
and P.N. Kumta, “Chemical Synthesis and Characterization
of Magnesium Substituted Amorphous Calcium Phosphate (Mg-ACP), Mat. Sci. Eng. C 30 (2010) 1313.
[35] J.L.
Moreau, L.M. Sun, L.C. Chow, and H.H.K. Xu, “Mechanical and Acid neutralizing
Properties and Bacteria Inhibition of Amorphous Calcium Phosphate Dental
Nanocomposite,” J. Biomed. Mater. Res.
B 98 (2011) 80.
[36] L. Pauling, General Chemistry,
Dover Publications, New York, 1988, p. 627.
[37] T.I. Ivanova, O.V. Frank-Kamenetskaya,
A.B. Koltsov, and V.L. Ugolkov,
“Crystal Structure of Calcium-deficient Carbonated Hydroxyapatite; Thermal
Decomposition,” J. Solid State Chem.
160 (2001) 340.
[38] L. Wang, I. Sondi, and E. Matijevic,
“Preparation of Uniform Needle-like Aragonite Particles by Homogeneous
Precipitation.” J. Colloid Surf. Sci.
218 (1999) 545.
[39] M.P. Staiger, A.M. Pietak,
J. Huadmai, and G. Dias, “Magnesium and Its Alloys as
Orthopedic Biomaterials: A Review,” Biomaterials
27 (2006) 1728.
[40] Y. Xin, K. Huo, H. Tao, G. Tang, and P.K.
Chu, “Influence of Aggressive Ions on the Degradation Behavior of Biomedical
Magnesium Alloy in Physiological Environment,” Acta Biomater. 4 (2008) 2008.
[41] X.N. Gu, W.R. Zhou, Y.F. Zheng, Y. Liu, and
Y.X. Li, “Degradation and Cytotoxicity of Lotus-type Porous Pure Magnesium as
Potential Tissue Engineering Scaffold Material,” Mater. Lett. 64 (2010) 1871.
[42] M. Tomozawa, S. Hiromoto,
and Y. Harada, “Microstructure of Hydroxyapatite-coated Magnesium prepared in
Aqueous Solution,” Surf. Coat. Tech.
204 (2010) 3243.
[43] J.Y. Uan, S.H. Yu, M.C. Lin, L.F. Chen, and
H.I. Lin, “Evolution of Hydrogen from Magnesium Alloy Scraps in Citric
Acid-added Seawater without Catalyst,” Int.
J. Hydrogen Energ. 34 (2009) 6137.
[44] S. Cazalbou, C. Combes, D. Eichert, C. Rey, and M.J. Glimcher,
“Poorly Crystalline Apatites: Evolution and
Maturation in vitro and in vivo,” J. Bone Miner. Metab. 22 (2004) 310.
[45] D. Bayraktar and A.C. Tas, “Chemical Preparation of Carbonated Calcium
Hydroxyapatite Powders at 37°C in Urea-containing Synthetic Body Fluids,” J. Eur. Ceram. Soc. 19 (1999) 2573.
[46] A.C. Tas, “Synthesis of Biomimetic Ca-Hydroxyapatite Powders at 37°C in Synthetic
Body Fluids,” Biomaterials 21 (2000)
1429.
[47] E. Landi, A. Tampieri,
G. Celotti, R. Langenati,
M. Sandri, and S. Sprio, “Nucleation of Biomimetic
Apatite in Synthetic Body Fluids: Dense and Porous Scaffold Development,”
Biomaterials 26 (2005) 2835.
[48] N. Nassif, F. Martineau, O. Syzgantseva, F. Gobeaux, M. Willinger, T. Coradin, S. Cassaignon, T. Azais, M.M. Giraud-Gille, “In
vivo inspired conditions to synthesize biomimetic hydroxyapatite,” Chem Mater. 22 (2010) 3653.
[49] C. Mossaad, M. Starr, S. Patil,
and R.E. Riman, “Thermodynamic Modeling of
Hydroxyapatite Crystallization with Biomimetic Precursor Design
Considerations,” Chem. Mater. 22
(2010) 36.
[50] C. Liu, Y. Huang, W. Shen, and J. Cui, “Kinetics of Hydroxyapatite
Precipitation at pH 10 to 11,” Biomaterials
22 (2001) 301.
[51] T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma,
T. Takeguchi, and W. Ueda, “Influence of Preparation
Factors on Ca/P Ratio and Surface Basicity of Hydroxyapatite Catalyst,” J. Jpn. Petrol.
Inst. 52 (2009) 51.
[52] D.G.A. Nelson, “The Influence of Carbonate on the Atomic Structure and
Reactivity of Hydroxyapatite,” J. Dent.
Res. 60C (1981) 1621.
[53] D.G.A. Nelson, G.J. Wood, J.C. Barry, and J.D.B. Featherstone, “The
Structure of (100) Defects in Carbonated Apatite Crystallites- A High
Resolution Electron Microscope Study,” Ultramicroscopy 19 (1986) 253.
[54] P. Koutsoukos, Z. Amjad,
M.B. Tomson, and G.H. Nancollas, “Crystallization of
Calcium Phosphates- Constant Composition Study,” J. Am. Chem. Soc. 102 (1980) 1553.
[55] L.W. Schroeder, B. Dickens, and W.E. Brown, “Crystallographic Studies
of the Role of Mg as a Stabilizing Impurity in b–Ca3(PO4)2. II. Refinement
of Mg-containing b–Ca3(PO4)2,”
J. Solid State Chem. 22 (1977) 253.
[56] J.D. Termine, R.A. Peckauskas,
and A.S. Posner, “Calcium Phosphate Formation in Vitro. II. Effects of Environment
on Amorphous-Crystalline Transformation,” Arch.
Biochem. Biophys. 140
(1970) 318.
[57] M. Harada, N. Udagawa, K. Fukasawa,
B.Y. Hiraoka, and M. Mogi, “Inorganic Pyrophosphatase Activity of Purified Bovine Pulp Alkaline
Phosphatase at Physiological pH,” J.
Dent. Res. 65 (1986) 125.
[58] R. Koncki, B. Rozum,
and S. Glab, “pH-metric Detection of Alkaline
Phosphatase Activity as a Novel Biosensing Platform,”
Talanta 68
(2006) 1020.
[59] T. Yabe, “The Effect of pH on Alkaline Phosphatase Activity in Serum of
the Rat and Other Species,” Arzneimittelforschung 35 (1985) 193.
[60] Y. Li, T. Wiliana, and K.C. Tam, “Synthesis
of Amorphous Calcium Phosphate using various types of Cyclodextrins,”
Mater. Res. Bull. 42 (2007) 820.
Table 2 Details
of select experiments
|
Experiment |
P source |
Ca source |
CO3 source |
P (mM) |
Ca (mM) |
CO3 (mM) |
Final pH |
Phases/XRD |
Medium |
|
1 |
-- |
Ca |
-- |
-- |
25 |
-- |
12.6 |
Ca(OH)2+CaCO3 |
H2O |
|
2 |
-- |
Ca |
NaHCO3 |
-- |
25 |
27 |
9.9 |
CaCO3 |
5 KCl+ 1.5 MgCl2 |
|
3 |
-- |
Ca |
NaHCO3 |
-- |
25 |
27 |
9.9 |
CaCO3 |
H2O |
|
4 |
-- |
Ca |
NaHCO3 |
-- |
25 |
27 |
12.3 |
CaCO3 |
95 NaCl |
|
5 |
-- |
Ca |
NaHCO3 |
-- |
25 |
27 |
12.3 |
CaCO3 |
MS |
|
6 |
Na2HPO4 |
Ca |
-- |
10 |
25 |
-- |
12.3 |
PCA+CaCO3 |
H2O |
|
7 |
Na2HPO4 |
Ca |
-- |
10 |
16.667 |
-- |
12.2 |
PCA |
MS w/o HCO3 |
|
8 |
Na2HPO4 |
Ca |
-- |
10 |
25 |
-- |
12.4 |
PCA |
MS w/o HCO3 |
|
9 |
(NH4)2HPO4 |
Ca |
-- |
10 |
16.667 |
-- |
11.3 |
ACP |
MS w/o HCO3 |
|
10 |
(NH4)2HPO4 |
Ca |
-- |
10 |
25 |
-- |
12.0 |
PCA |
MS w/o HCO3 |
|
11 |
Na2HPO4 |
CaCl2×2H2O |
-- |
10 |
25 |
-- |
5.9 |
DCPD+PCA |
H2O |
|
12 |
(NH4)2HPO4 |
CaCl2×2H2O |
-- |
10 |
16.667 |
-- |
6.5 |
DCPD |
MS w/o HCO3 |
|
13 |
(NH4)2HPO4 |
CaCl2×2H2O |
-- |
10 |
25 |
-- |
6.5 |
DCPD+PCA |
MS w/o HCO3 |
|
14 |
(NH4)2HPO4 |
CaCl2×2H2O |
-- |
10 |
50 |
-- |
5.7 |
DCPD+PCA |
MS w/o HCO3 |
|
15 |
(NH4)2HPO4 |
CaCl2×2H2O |
-- |
10 |
16.667 |
-- |
6.1 |
DCPD+PCA |
H2O |
|
16 |
Na2HPO4 |
Ca |
NaHCO3 |
1 |
2.5 |
27 |
9.2 |
ACP |
MS |
|
17 |
Na2HPO4 |
Ca |
NaHCO3 |
5 |
12.5 |
27 |
10.3 |
ACP |
MS |
|
18 |
Na2HPO4 |
Ca |
NaHCO3 |
10 |
25 |
27 |
12.0 |
ACP+CaCO3 |
MS |
|
19 |
Na2HPO4 |
Ca |
NaHCO3 |
10 |
25 |
27 |
9.0 |
No ppts |
H2O |
|
20 |
(NH4)2HPO4 |
Ca |
NaHCO3 |
10 |
25 |
27 |
10.4 |
ACP+CaCO3 |
MS |
|
21 |
Na2HPO4 |
Ca |
NH4HCO3 |
10 |
25 |
27 |
10.1 |
ACP+CaCO3 |
MS |
|
22 |
(NH4)2HPO4 |
Ca |
NH4HCO3 |
6.667 |
16.667 |
27 |
9.4 |
ACP |
MS |
|
23 |
(NH4)2HPO4 |
Ca |
NH4HCO3 |
10 |
16.667 |
27 |
9.3 |
ACP |
MS |
|
24 |
(NH4)2HPO4 |
Ca |
NH4HCO3 |
10 |
25 |
27 |
9.5 |
ACP |
MS |
|
25 |
Na2HPO4 |
CaCl2×2H2O |
NaHCO3 |
10 |
25 |
27 |
7.0 |
PCA |
MS |
|
26 |
Na2HPO4 |
CaCl2×2H2O |
NaHCO3 |
10 |
25 |
27 |
7.0 |
PCA |
H2O |
|
27 |
(NH4)2HPO4 |
CaCl2×2H2O |
NH4HCO3 |
10 |
25 |
27 |
7.0 |
PCA |
MS |
|
28 |
Na2HPO4 |
Ca-acetate |
NaHCO3 |
10 |
25 |
27 |
7.0 |
PCA |
MS |
|
29 |
Na2HPO4 |
Ca-nitrate |
NaHCO3 |
10 |
25 |
27 |
7.0 |
PCA |
MS |
|
30 |
Na2HPO4 |
Ca(OH)2 |
NaHCO3 |
10 |
25 |
27 |
11.7 |
ACP+CaCO3 |
MS |
Figure
Captions (see all the below Figures in http://www.cuneyttas.com/Ca-metal.pdf)
Fig 1a XRD
traces of as-received Ca granules (bottom) and Ca granules stirred in H2O
or MS (top, Exp. 1)
Fig
1b FTIR traces of the samples of experiments
3, 4, and 5
Fig
1c Macrophotograph
of as-received Ca metal granules (shots)
Fig
1d SEM photomicrograph of the CaCO3
samples of experiment 5
Fig 2 pH-time
curves for experiments 2, 3, 4, and 5 (the moment of dissolution of Ca granules
were indicated for experiments 4 and 5)
Fig 3a XRD traces of
the samples of experiments 6, 7, 8, 9, and 10 (solution pH values, at the end
of 25 min of stirring, were shown on the traces)
Fig 3b FTIR traces of
the samples of experiments 1, 6, 7, 8, and 9
Fig 3c pH-time curves
for experiments 1, 5, and 8 (the moment of dissolution of Ca granules was
indicated by the arrows for experiments 5 and 8)
Fig 4 Combined XRD
and FTIR traces for the samples of experiments 6 and 11 (the bottom XRD trace
for PCA of experiment 6, the XRD trace for DCPD of experiment 11 shown on top)
Fig 5a Combined XRD
and FTIR traces for the samples of experiments 16, 17, and 18 (CaCO3
peaks were indicated by + in the XRD trace of experiment 18)
Fig 5b pH-time curves
for experiments 16, 17, 18, and 19 (the dissolution time of Ca granules was
indicated by the straight dashed line)
Fig 5c SEM photomicrograph of the sample
of experiment 16
Fig 5d SEM photomicrograph of the sample
of experiment 18
Fig 6 Combined XRD
and FTIR traces for the samples of experiments 22, 23, and 24
Fig 7 Combined XRD
and FTIR traces for the samples of experiments 25 through 29
Fig 8a XRD traces of
the samples of experiments 18, 20, 21, and 30
Fig 8b FTIR traces of
the samples of experiments 18, 20, 21, and 30
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}